

The last two years have seen significant changes in the approval management of clinical trials. In the UK, the Combine Review system became mandatory since the 1st January 2022 and in the European Union, the previous Directive has been replaced by the EU Clinical Trials Regulation since the 31st January 2023.
These two systems shared comparable goals in their efforts to revitalize the appeal of conducting research studies in the UK and EU. Their aim is to improve efficiency and speed in the approval process and to establish a consistent and harmonized set of documentation requirements.
They also increase transparency by disseminating research findings to a broader audience, including both the research community and patients. Additionally, in the EU, documents submitted as part of the Clinical Trial Application (CTA) will eventually be made public.
It is important to note that all these improvements must be implemented while maintaining the highest safety standards for all participants involved in the studies. Learn more below.
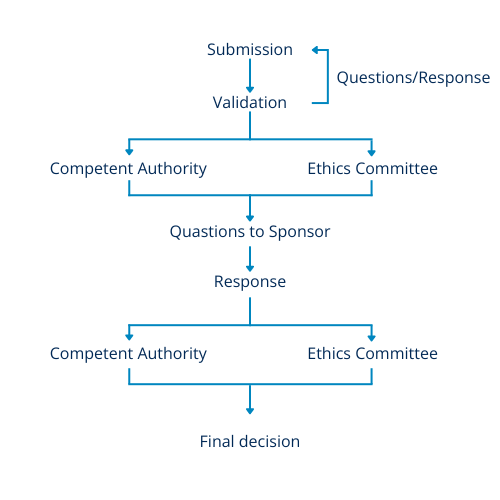
Figure 1 – Overview of the CTA
Overview of the CTA process: A Comparative Glance
A Unified Submission Process
At a high level, both the UK and EU systems for clinical trial submissions share similarities. A single submission is made to both the Competent Authority (CA) and Ethics Committee (EC) through a centralized portal, eliminating the need for separate submissions with overlapping documents. However, this also means that the submission cannot be made until all necessary documents are completed.
In the UK, the submission is processed through the IRAS portal, while in the EU, it is handled by the CTIS portal hosted by the European Medicines Agency (EMA). The EMA, however, is not involved in clinical trial assessments.
Streamlining the Documentation
Standardized core documentation is used across all EU countries for scientific documents, and each country has a templated list of required documents, eliminating additional local requirements. One critical decision is thus selecting the country that will lead the study assessment, which will be reviewed and commented by the other countries. The most popular countries for this will be discussed later.
The CA evaluates the scientific aspects of the submission, including chemistry, nonclinical, and clinical data, to assess overall benefit-risk. Meanwhile, the EC reviews patient-facing materials such as informed consent and recruitment materials and evaluates the site and investigator’s competency in protecting participants.
Stages and Timelines
In both systems, a single set of questions is received, and a single set of responses is submitted. Figure 1 offers a summary of the two processes utilized in both the UK and EU for reviewing clinical trial applications. The first stage is an initial validation check to ensure that all necessary documents are included. Following this, the documents are made accessible to the appropriate regulatory bodies for review.
Questions that arise during the review process are gathered and presented as a single set to the sponsor. However, in the UK, attending the meeting with the Ethics Committee (EC) may provide insight into the potential questions. The sponsor then provides responses to all questions through the online portal, and the information is distributed for final decision-making.
The differences between the two systems arise when looking at the timelines of the CTA process. It is easily explained by the fact that the EU systems must accommodate submissions to multiple countries. The timelines shown below represent the maximum allowable time and can be shorter on a case-by-case basis.
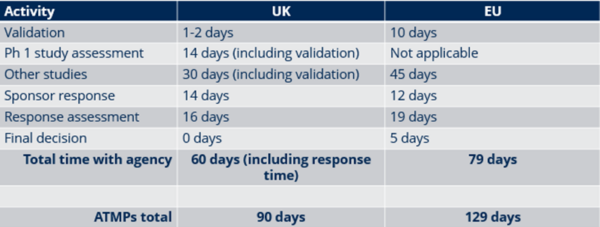
It is worth noting that the UK continues to prioritize Phase 1 studies due to historical reasons. In the past, studies involving healthy volunteers were not regulated by the MHRA. When the Directive was introduced nearly 20 years ago, Phase 1 studies became regulated by the MHRA and were granted a shorter review time to maintain the UK market’s appeal for these studies.
MHRA's Recent Announcements
On 21st March this year, a press release was issued by the MHRA, announcing, amongst things, plans to enhance the UK’s competitiveness and incorporate the Combined Review process into law. The MHRA intends to adopt a risk-based approach to different types of studies to make their requirements more proportional to the level of risk involved. In line with the EU, the MHRA has also made it mandatory to publish the results of clinical studies, which was previously only encouraged but not a legal requirement.
Deciphering Clinical Trial Metrics
In the context of Clinical Trials Application, metrics refer to quantitative measurements used to assess various aspects of the application process. They are used to monitor the efficiency and effectiveness of the application process and to identify areas for improvement. They also provide transparency and allow stakeholders to track progress, observe trends, and make data-driven decisions.
EU's Commitment to Transparency
EMA is publishing monthly metrics on the Clinical Trials regulation, which demonstrates the EU’s commitment to transparency. These metrics cover various aspects, such as the number of applications, therapeutic areas involved, development phases of studies and the countries involved in the study. In total, there are 18 metrics.
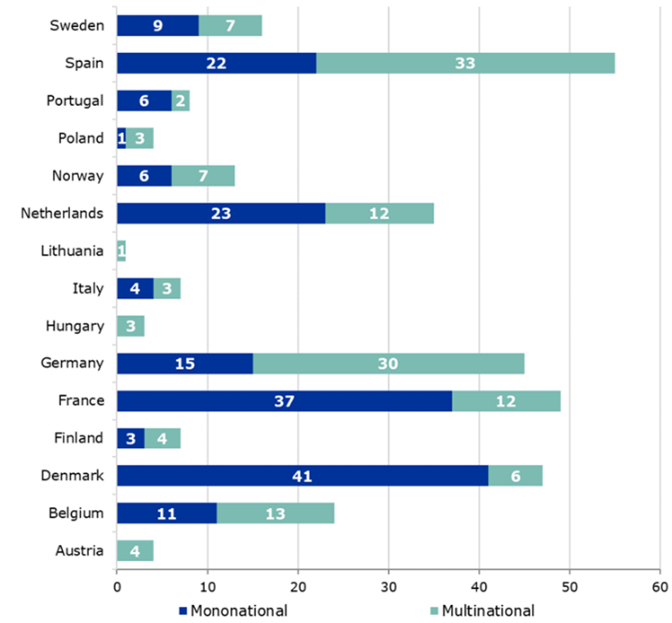
Figure 2 – Countries chosen to lead the assessment
The metric on the figure above is particularly interesting as it shows which countries sponsors have been selected to lead the application, with the dark blue indicating the number of studies being performed in that country.
It is worth noting that some countries have larger populations and are therefore more attractive for recruitment purposes. Countries such as Denmark, the Netherlands and Belgium, are specialized in early-phase studies and offer quicker review timelines than the standard timelines.
The EMA also provides data on the time taken for each clinical trial application to reach a decision, which is an important metric for assessing the efficiency of the application process. While this data is not publicly available in the form of a graph, we have prepared one based on the first year of the Regulation being in operation.
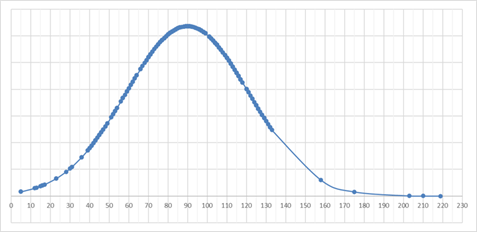
Figure 3 – Number of calendar days from submission to the time of the first decision
The graph above shows the distribution of the time to make decision for each application, with each point representing a single application. It is worth noting that advanced therapy medicinal products (ATMPs) are included in this dataset and are likely at the right end of the curve.
It is encouraging to see that some studies were approved in a matter of days, which may be due to prior interactions with regulatory authorities through scientific advice. Overall, most applications are able to achieve the published timelines.
UK's Performance Overview
In the UK, for many years, the MHRA has been publishing metrics on its performance against target. However, the last set of metrics were published in September 2022 and they only cover data up until March 2022, which means that any recent data on the time to approval are not available. At that time, they were meeting the target of 14 days for Phase 1 studies and 30 days for other studies.
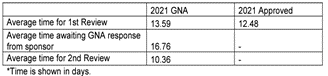
Figure 4 – Phase 1 performance
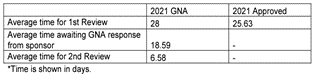
Figure 5 – Phase 2-4 performance
Unfortunately, the timelines now are not currently being met due to resourcing issues at the MHRA. The MHRA has acknowledged the current situation and is taking steps to address the issue by triaging applications and recruiting additional nonclinical and medical assessors, to solve this issue.
Practical Insights for Clinical Study Management
It has been noted that sponsors may tend to take a conservative approach when deciding whether to submit a substantial amendmen t, which was previously manageable when the MHRA had sufficient resources to handle the workload.
However, considering the current situation where the published timelines are not being met due to resourcing issues, it may be necessary to revisit the first principles that state that the responsibility to assess whether an amendment is ‘substantial’ lies with the sponsor.
Care must be taken to avoid over-reporting, as it can be resource-intensive for all involved and may lead to delays in the study’s progression. Therefore, it is advised that the sponsor involves all relevant parties in the decision-making process and carefully considers whether an amendment is truly substantial before submitting it.
It is important to ensure that any decision regarding whether a change is substantial or non-substantial is well-documented and filed in the Trial Master File. Indeed, such documentation may be subject to review during any Good Clinical Practice (GCP) inspection.
Moreover, to ensure the accuracy of these decisions, it is recommended that the regulatory team is involved in the process and that they closely review any changes against the relevant guidelines and specific examples provided in the protocol, IMPD, IB updates, etc. Only changes that have a significant impact on the study should be submitted as substantial amendments.
When the next substantial amendment is required, any non-substantial amendments that have occurred in the interim should be collated, clearly identified as such in the cover letter, and included in the application to bring the CTA up to date.
Wrapping Up: The Road Ahead for Clinical Trials
There have been substantial changes in clinical trials regulations in both the UK and EU, with more changes to come. Creating high-quality submissions can save resources for competent authorities, ECs, and sponsors by minimizing the need for follow-up questions. It is important for sponsors to make informed decisions about whether an amendment is substantial or not, and involve relevant parties in the decision-making process.

with ease.

Navigate Regulatory Complexity with Confidence
Navigate complex regulatory landscapes with expert guidance across pharmaceuticals, medical devices, and IVD.
Read more

