
In earlier blog posts, we spotlighted the EU member states – Germany, Belgium, and France – and their adoption of the fast-track procedure for reimbursement.
Now, let’s delve into the intricacies of Digital Therapeutics (DTx) regulation in the UK, especially in light of Brexit in January 2020.
We’ll cover UKCA and CE marking, NHSx and DTAC requirements, as well as DTx reimbursement in the UK.
UK’s Post-Brexit Regulatory Landscape
Post-Brexit, DTx products in the UK are regulated as medical devices, necessitating either a CE or UKCA mark.
-
UKCA Marking: The regulatory requisites for medical devices in the UK are encapsulated within the UK law MDR 2002 (amended). Under this law, DTx, being a medical device, must fulfil all MDR 2002 requirements to secure a UKCA marking from approved UK bodies.
-
CE Marking: In light of the economic implications post-Brexit, the UK opted for a gradual transition for medical device regulations, extending significant transition periods. Consequently, CE-marked products will continue to be accepted in the UK at least until 30 June 2030, depending on device classification. Comprehensive details on transitional periods can be located on the MHRA website. Further insights into CE-marked DTx can be gleaned from the previous chapter.
The Role of NHSx and DTAC in UK’s DTx Landscape
Beyond the CE or UKCA markings, DTx products in the UK undergo evaluations by NHSx and NICE, ensuring their economic viability, reimbursement potential, and accessibility for patients in line with the UK’s regulatory framework.
NHSx is a collaborative initiative between the Department of Health and Social Care, NHS England, and NHS Improvement. Its inception was driven by the aspiration to digitally transform care, a vision spearheaded by England’s Transformation Directorate from February 2020 onwards.
In the subsequent month, NHSx unveiled the Digital Health Technology Standard, which elucidates the requisite benchmarks that digital health technology must attain within the UK. This standard, also known as the Digital Technology Assessment Criteria (DTAC), encompasses domains like clinical safety, data protection, technical security, interoperability, and usability & accessibility.
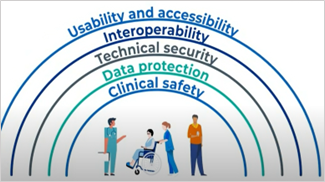
Figure 1 – Key components of DTAC assessment criteria ( Source)
The DTAC requirements are intrinsically tied to the UK’s health IT clinical risk standard, known as DCB0129. This standard guides the development and maintenance of Digital Therapeutics (DTx). Furthermore, DTAC incorporates stipulations from the UK’s Medical Device Regulation (MDR) 2002, albeit with amendments.
To ensure a comprehensive assessment for UK reimbursement approvals, DTAC has also introduced specific criteria that cater to five fundamental pillars: Clinical Safety, Data Protection, Technical Security, Interoperability, and Usability & Accessibility.
An accessible self-assessment form aids stakeholders in ensuring their digital solutions align with UK standards, and interested parties can even subscribe for email notifications about impending DTAC modifications.
The Path to DTx Reimbursement in the UK
NICE, as the Health Technology Assessment Agency, lays down the evidence framework. It has formulated 21 different evidence standards ( ESF) to equip the MHRA with tools for evaluating health technologies. These standards also guide digital health tech innovators and manufacturers proactively. DTx designers and developers can contact NICA and MHRA in an early stage.
Depending on the technology’s intended use, it’s assigned to one of the three tiers which determine its evidence requirements:
-
Tier A: Technologies aimed at cost-saving or freeing up staff time without direct patient or health outcomes.
-
Tier B: Technologies assisting individuals in managing their health and well-being.
-
Tier C: Technologies meant for diagnosing, treating medical conditions, or guiding care choices.
Should a product receive a favorable evaluation by NICE, indicating benefits for both patients and the NHS, it becomes eligible for the reimbursement program.

Figure 2 – Overview of NICE evidence standards vs. the pertinent ESF Digital Health Technology risk level ( Source)
Conclusion
While the UK might initially appear as a complex market for DTx and digital health entities, the streamlined processes like CE and UKCA markings, combined with specific guidelines like DTAC and NICE evidence standards, make it an appealing destination.
The UK’s proactive approach, emphasizing early engagement between DTx developers and national evaluation (NICE) and approval (MHRA) entities, underscores its commitment to enhancing healthcare outcomes while optimizing costs.
In the next DTx blog post, we’ll transition from the European regulatory milieu to explore what the vast regulatory landscape of the United States holds for DTx.

Navigate Regulatory Complexity with Confidence
Navigate complex regulatory landscapes with expert guidance across pharmaceuticals, medical devices, and IVD.
Read moreSubscribe to the latest updates in life science
Expert perspectives delivered to your inbox — pick your interests.
No spam, ever. Unsubscribe anytime.


