

An Investigational Medicinal Product Dossier (IMPD) is a critical document required for clinical trial applications (CTAs) in the European Union and the United Kingdom. It is governed by Regulation (EU) No 536/2014 in the EU and by The Medicines for Human Use (Clinical Trials) Regulations 2004 SI 2004 No. 1031 in the UK. The IMPD is generally organized into 4 primary sections:
- Quality
- Non-clinical
- Clinical
- Safety and efficacy
In this blog post, we focus on the key considerations for the Quality section of the IMPD for chemically defined investigational medicinal products (IMP).
After months of work on your CTA, ensuring your IMPD meets quality standards sets you up for success and avoids unnecessary delays. Common issues like insufficient justification of specifications or missing batch analysis data can lead to regulatory Requests for Information (RFIs), adding time and complexity to the review process.
Having worked on over 100 CTAs across the EU and UK, we’ve supported numerous companies in navigating these challenges and maintaining regulatory compliance. Preparing a thorough, compliant IMPD from the start helps avoid these setbacks, ensuring smoother progression to clinical phases and keeping your trial on track.
What is the IMPD quality section?
The IMPD quality section must provide comprehensive documentation on the chemical and pharmaceutical quality of the IMPs. This covers aspects like Chemistry, Manufacturing, and Controls (CMC), stability data and packaging.
The Quality IMPD structure aligns with the Common Technical Document (CTD) format, particularly CTD Module 3: Quality. The module headings in Figure 1 are used when writing the Quality IMPD for Drug Substance and Drug Product.
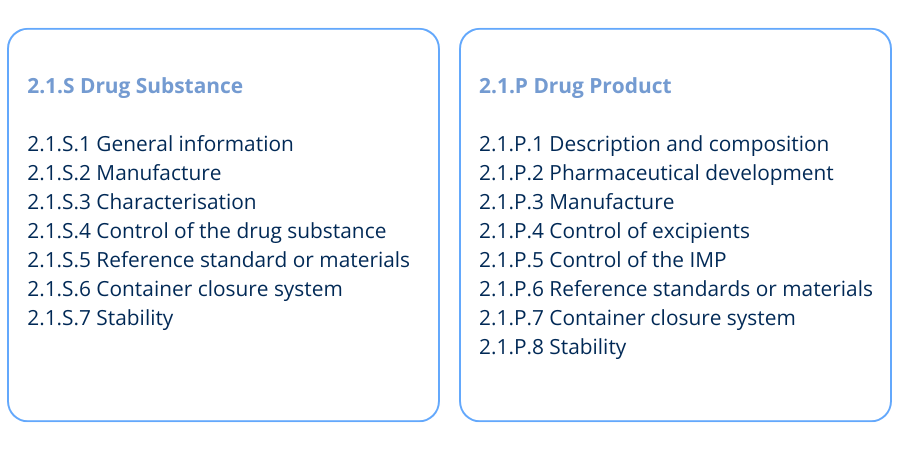
It’s also important to consider how requirements evolve throughout clinical development. In Phase I, the focus is on safety, with limited manufacturing and characterisation data.
By Phase III, however, regulators expect detailed information on stability, specifications, and comprehensive batch data (see Figure 2). This makes phase-specific planning essential for sponsors working with IMPDs in clinical trials.

Table 1 - Increasing Regulatory Requirements for IMPD Quality Section from Phase I to Phase III
Key quality considerations for IMPD Clinical Trials
Below is a summary of key considerations for the requirements of each IMPD section. These are based on the common deficiencies identified by the Medicines and Healthcare products Regulatory Agency (MHRA) when assessing CTAs as well as QbD’s extensive experience in this area.
1. Characterisation data
Missing, inadequate, or poorly interpreted characterisation data is a frequent issue. Regulators typically expect the drug substance structure to be confirmed by at least two spectroscopic methods, with representative spectra included in the IMPD.
2. Method validation data
The IMPD must include a summary of analytical method validation for Phase II and III trials. In Phase I, confirmation of method suitability is sufficient. Inadequate summaries can result in deficiencies.
3. Batch analysis data
These data demonstrate that each manufacturer can consistently produce the drug substance and product at the proposed scale(s) throughout clinical development. Data must be included for all listed manufacturing sites and inclusion of drug substance batches used for non-clinical studies should not be overlooked. Missing this information can trigger RFIs.
4. Justification of specifications
Sponsors must justify specifications, acceptance criteria, and control methods. Impurity limits should be backed by safety/toxicity data and batch data. If expected characteristics are not controlled, this must be justified.
5. Retest period
Stability data should be summarised in table form, showing that the drug substance meets specifications at manufacture. The re-test period should be based on this data. Missing or insufficient stability data often results in RFIs.
6. Shelf life
The proposed shelf life (IMP) must be supported by stability data, although extrapolation can be usedto set this. A shelf-life extension plan and justification criteria must also be included. This is a common deficiency flagged by agencies.
7. TSE/BSE certification
Sponsors must include documentation controlling for transmissible spongiform encephalopathy (TSE) agents, such as certification and relevant process controls. Missing or incomplete data will result in RFIs.
8. Qualified Person (QP) declarations
Sites manufacturing IMP that are located outside of the EU/EEA require a QP declaration confirming EU GMP compliance. This will require an on-site GMP audit, so planning ahead is crucial. Incomplete declarations are a frequent source of delays.
9. Labelling
CTA submissions must include labelling samples for all IMPs, including comparators, in line with Annex 13 of EU GMP guidelines. Any deviations must be justified. Unjustified deviations often require clarification or amendment.
10. GMP Compliance
The IMPD must be supported by GMP documentation such as the Manufacturing and ImportationAuthorisation (MIA) and QP certification (Annex I, Section G, Clinical Trial Regulation (CTR). MIA is required for all manufacturing, packaging, and labelling sites.
Common IMPD mistakes to avoid in Clinical Trials
Transitioning between regulatory frameworks, such as from the IMPD to an IND (Investigational New Drug) application, can be challenging due to differences in content, format, and requirements. Comparing the two and maintaining alignment is critical.

Table 2 - Key regulatory differences when transitioning between IMPD and IND submissions
Writing the IMPD quality section efficiently
A well-written IMPD Quality section can save time, prevent RFIs, and support smoother clinical trial progress. The following strategies help ensure your documentation is both compliant and efficient:
- Adhering to guidelines: Follow the European Commission's Guideline on the Requirements for the Chemical and Pharmaceutical Quality Documentation.
- Automation and templates: Streamline your IMPD preparation process with internal templates.
- Versioning: Maintain strong version control to reflect ongoing changes. This is particularly important for IMPD clinical trials involving multiple studies or regions.
Why work with experts for IMPD Clinical Trials?
It’s highly recommended to involve an IMPD subject matter expert before submission. They can help proactively identify and resolve potential gaps, ensuring compliance and minimizing delays.
QbD has supported numerous companies in obtaining and maintaining compliant CTAs in the EU and UK. Our expert team can assist with authoring, reviewing, and maintaining IMPDs in clinical trials, as well as responding to agency queries. With up-to-date regulatory intelligence and a database of common deficiencies, we ensure a smooth submission process.
Need support with your IMPD?
Reach out to QbD’s regulatory affairs experts today. We’ll help you prepare a strong, compliant IMPD so you can focus on moving your clinical trial forward with confidence.
Let’s talk about how we can support your team.

References
1.EUR-Lex - 02014R0536-20221205 - EN - EUR-Lex [Internet]. eur-lex.europa.eu.
The Medicines for Human Use (Clinical Trials) Regulations 2004 [Internet]. Legislation.gov.uk. 2015.
UK Government. Common issues identified during clinical trial applications.
European Commission. Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials.
About the Author

Specialist RA Pharma at QbD Group
Stella specializes in Regulatory Affairs for pharmaceuticals at QbD Group, supporting companies with regulatory submissions and compliance strategies.

Navigate Regulatory Complexity with Confidence
Navigate complex regulatory landscapes with expert guidance across pharmaceuticals, medical devices, and IVD.
Read more

