

1. From idea to requirements: kickstarting design & development
Start with your intended use and user needs. What problem does your software solve for patients or clinicians? Once that’s clear, translate those needs into system and/or software requirements—this is where your actual development process begins.
To align with global expectations, especially in Europe and the U.S., get familiar with these key standards:
- IEC 62304 – The core standard for medical software development and lifecycle
- IEC 82304 – Focused on software validation for standalone MDSW.
- ISO 14971 – The gold standard for risk management in medical devices.
- IEC 62366 – Usability engineering, crucial for user interface design.
Together, these standards shape your development process, validation approach, and technical documentation.
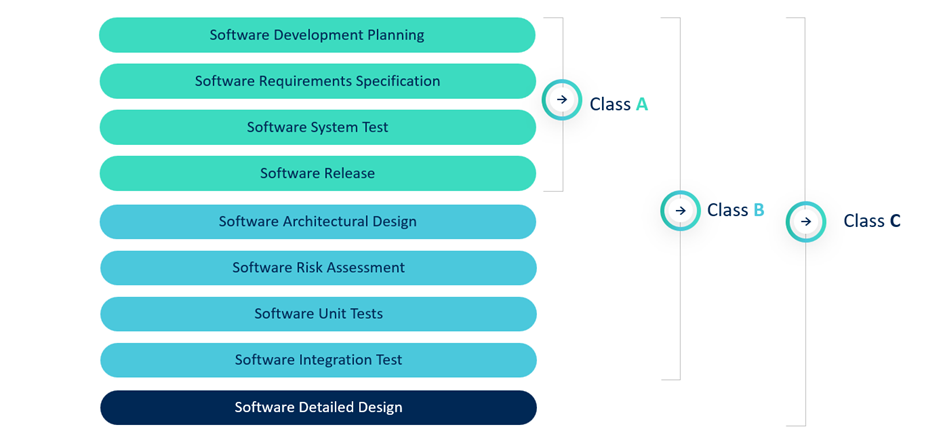
Safety classification: A, B, or C?
IEC 62304 classifies software in 3 safety classes:
- Class A: No risk of injury (rare for MDSW).
- Class B: Risk of non-serious injury.
- Class C: Risk of serious injury or death.
Most start-ups fall into Class B. The classification dictates the required deliverables—for instance, Class C must include a detailed design, while Class A has fewer documentation requirements.
Important note: Notified bodies rarely accept Class A without a robust, well-justified rationale. As highlighted in recent QbD webinars, even minor functionalities can contribute to risk, making Class B the more defensible default for most start-ups.
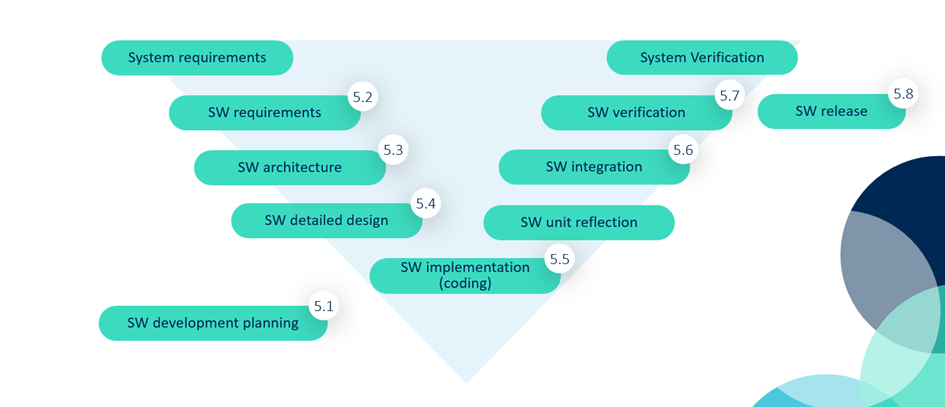
What does IEC 62304 clause 5 actually look like?
IEC 62304 outlines the required steps for software development under Clause 5. Here’s a simplified walkthrough of what’s expected, especially for Class B or C software:
5.1 – Software Development Planning
Define how you’ll approach development, testing, risk management, and change control. This is your roadmap.
5.2 – Software Requirements Analysis
Translate system and user needs into precise software requirements.
5.3 – Software Architecture Design
Outline the high-level structure of your software (components, interfaces, data flow, etc.).
5.4 – Software Detailed Design (Class C only)
Dive deeper into unit-level design, enabling accurate implementation and testing.
5.5 – Software Unit Implementation and Testing
Write code and test each unit (e.g., through code review, automated tests, or static analysis).
5.6 – Software Integration and Integration Testing
Combine units and verify they work together as intended.
5.7 – Software System Testing (Verification)
Make sure the complete system meets all software requirements.
5.8 – Software Release
Document and control the final version. Confirm readiness for market or clinical use.
You can visualize this lifecycle using the so-called V-model, where each development activity on the left has a corresponding verification step on the right:
IEC 62304 software lifecycle visualized as a V-model—each development phase aligns with a verification step.
This structure not only guides your development and testing, but it also shapes your software technical file. Notified bodies will expect clear documentation aligned with this lifecycle.
2. Agile vs. waterfall: choosing your development model
IEC 62304 is process-oriented, not methodology-specific. That means you can use Waterfall, Agile, or a hybrid approach, as long as you document your process and meet the required deliverables.
Waterfall
- Clear, sequential stages
- Easier traceability
- Slower and less flexible
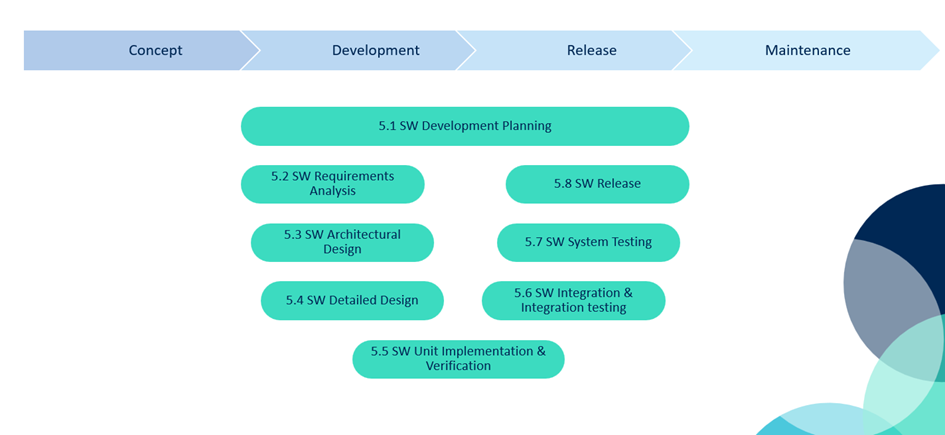
Waterfall maps well to IEC 62304, especially for projects with fixed requirements and longer timelines. As shown above, each phase follows the next, and testing is concentrated toward the end.
Agile
- Rapid iterations and sprints
- Shorter release cycles
- Requires strong documentation discipline to remain compliant
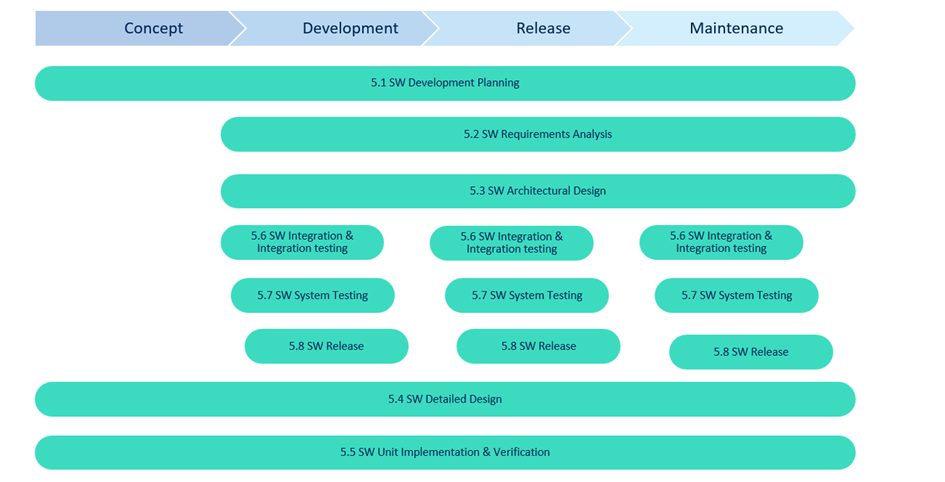
Agile, on the other hand, emphasizes flexibility, with overlapping activities and ongoing testing. IEC 62304 doesn’t prohibit Agile, but staying compliant requires discipline in documentation, especially traceability and risk management.
Best of both worlds: the hybrid model
For start-ups, a hybrid model often works best. You maintain high-level lifecycle documents (e.g. development plan, risk management) and update testing and code-related documentation sprint by sprint. This lets you stay flexible while keeping compliance in check.
Total agile development is often difficult to achieve. But the combined agile and waterfall approach, with some lifecycle documents and smaller agile cycles, can lead to releases with shorter lead times.
When using a more agile development approach, take into consideration that certain modifications to your software will require a notified body review before release. This can impact a fully agile release schedule and underlines the need for a well-defined release strategy so that business needs match regulatory compliance.

3. Avoiding compliance pitfalls
Even with the right model, early-stage companies can get tripped up on compliance details. Here are three areas to watch:
Risk management
ISO 14971 applies to both product and process, with patient safety and security risk definitions.
Start-ups must:
- Identify and evaluate risks
- Define risk controls
- Show traceability from risk controls to their effectiveness check.
SOUP (Software of Unknown Provenance)
Using (open source) libraries? That’s SOUP. You’ll need to:
- Justify use via risk analysis
- Monitor for vulnerabilities
- Track versions and updates
SOUP is assumed to produce more faults than Class B or C code. That’s why rigorous risk analysis, monitoring, and justification are key.
Configuration & change management
Start-ups often lack formal systems here, but it’s essential to:
- Version-control your codebase and documents (e.g. Git)
- Track changes with rationale
- Keep old versions accessible
Tip: Document your change control and bug-handling procedures from the start. It will save headaches when something breaks, or a notified body starts asking questions.
4. Getting ready for the AI Act and cybersecurity demands
The AI Act
If your MDSW uses AI or machine learning, the EU AI Act will apply from August 2027 for medical devices, but some requirements are already in force. It introduces new requirements, including:
- Data governance (training data acquisition, labeling)
- Logging and human oversight
- Robustness, accuracy, and cybersecurity checks
Start by performing a gap assessment between your current processes and AI Act expectations. Many requirements overlap with MDR/IVDR, but not all.
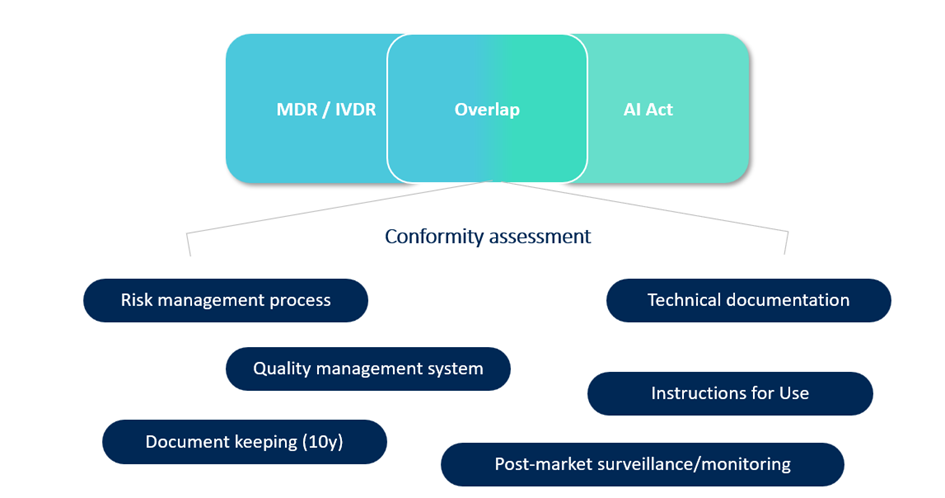
As shown above, some foundational elements—like risk management, documentation, and post-market monitoring—are familiar from both MDR/IVDR and the AI Act but still might require some updates. But new layers such as data governance, human oversight, and AI-specific logging may require additional controls.
You’ll need to manage three types of risk: patient safety (MDR/IVDR), cybersecurity, and now fundamental rights (AI Act).
Cybersecurity
This is now a top priority for notified bodies. Requirements include (among others):
- Cybersecurity risk analysis (aligned with ISO 14971)
- Penetration testing and threat mitigation
- Vulnerability monitoring (especially for SOUP components)
Even though ISO/IEC 81001 isn’t harmonized yet, it's smart to start aligning with its expectations. Cyber threats aren’t waiting for regulations to catch up.
Looking at a submission with the FDA? Make sure to keep the FDA guideline, “Cybersecurity in Medical devices” in mind, which will provide you with detailed information about the FDA’s expectations.
5. What Notified Bodies expect
Start-ups often think "lean" means "light on documentation." Unfortunately, notified bodies don’t agree. When reviewing your file, clear and easy-to-understand documentation on the processes you followed and deliverables you created go a long way.
Conclusion: start smart, stay compliant
Developing medical device software as a start-up is tough. But it’s absolutely doable with the right foundation.
- Use IEC 62304 and ISO 14971 as your development backbone.
- Choose a model that fits your team’s speed and strengths.
- Start small but smart: track your risks, manage your SOUP, and document decisions early.
- Don’t underestimate cybersecurity and AI regulation—future-proof now, not later.
- Build lean but acceptable documentation for your software class.
QbD Group is here to support you every step of the way, from shaping your software strategy to preparing for audits. Whether you’re still building your MVP or gearing up for CE marking, we can help you move faster without cutting corners.
Let’s turn your innovative software into a safe, compliant, and successful medical device.
Contact us for expert guidance.
About the Author

Project Manager at QbD Group
Pieter is a Project Manager at QbD Group, coordinating multi-disciplinary teams to deliver quality and regulatory consulting projects.


