













Types of Inspections
The objective of routine PV inspections is to assess whether the MAH can identify, characterise and report new or changed risks associated with their medicinal products.
Routine inspections can be either initial or re-inspections and are scheduled as part of the MHRA’s national inspection program according to a risk-based approach in line with the Good Pharmacovigilance (GVP) Module III. The MHRA will provide notice to the MAH before the inspection for preparedness.
Depending on the findings, the MAH may be subject to a re-inspection, otherwise, if there are no critical findings, there is no specific timeline by which an MAH must be inspected again, and this will be determined using a risk based approach.
Triggered inspections, also known as ‘for cause inspections’ can be scheduled due to intelligence or a previous critical finding. This includes if the MHRA have been informed of specific risk information regarding the MAH and its authorised products. The following breaches of the GPvP also call for a triggered inspection:
-
a whistleblower
-
other MHRA departments
-
another regulatory authority
For these types of inspections, very little or no notice is given in advance.
MHRA GPvP Inspection Metrics for 2022/2023
There were 17 PV inspections conducted in 2022/2023 which included routine inspections and triggered inspections.
A significant number of inspections conducted during this period were ‘for cause – intelligence’ inspections. The graph below shows the breakdown of findings issued by the MHRA along with the inspection type.

Source: Medicines and Healthcare products Regulatory Agency

Source: Medicines and Healthcare products Regulatory Agency
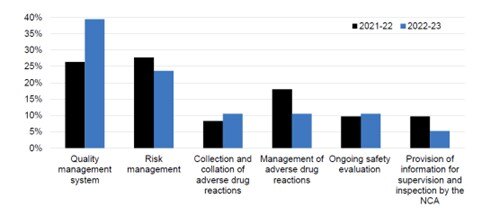
Source: Medicines and Healthcare products Regulatory Agency
The GPvP team at the MHRA continue to improve their process of how inspections are conducted to ensure that it is time and cost-effective for all parties. It is crucial to ensure that the foundations, such as a QMS are in place for an MAH to establish and maintain the PV activities necessary for a fully compliant and risk-based approach for medicinal products.
Our Vigilance department at QbD Group will collaborate closely with you to understand your business needs and objectives and then offer specialised solutions to assist you in accomplishing them. With our expert guidance, you can confidently navigate pharmaceutical regulations.



%20Checklist.jpg)







.jpg)




.jpg)
.jpg)

.jpg)


.jpg)
.jpg)
.png)