

The EU Medical Device Regulation 2017/745 (MDR) and EU In Vitro Diagnostic Regulation 2017/746, (IVDR)bring new obligations with them. One of them is the UDI system. In this blog post, you will learnwhat theUDI systemis and – more importantly – whyyou should start your own UDI process in time.
What is the consequence of UDI for manufacturers?
The Unique Device Identifier assigns a unique barcode to each individual medical device on the European market. All medical device manufacturers will have to apply the UDI, which will allow importers and distributors to use it for traceability purposes. And regulators, patients, doctors, and others will have easy access to the product information because all UDIs will be recorded in the EUDAMED database.
As a manufacturer, you are responsible for
- the UDI assignment
- the registration in the EUDAMED database
- the integration of the UDI carrier on the label or packaging of the device ( read more about UDI for MDSW here)
The implementation of the UDI system could be challenging. Therefore, we have gathered all relevant information to create a clear overview of the specific UDI requirements.
What exactly is a UDI?
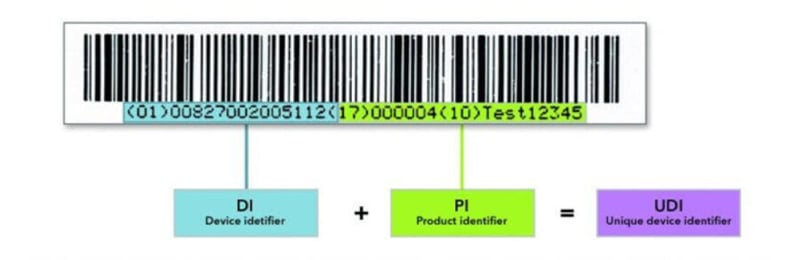
A UDI is a series of numeric or alphanumeric characters specific for a device and manufacturer and comprises the UDI-DIand the UDI-PI.
A UDI-DI(Device Identifier) is required for each individual product and is specific to a version or model of a device, providing access to the information listed in Annex VI Part B. The UDI-DI is therefore static.
The UDI-PI stands for the UDI Production Identifier and identifies the device production unit and, if applicable, the packaged devices, as specified in Annex VI Part C. The UDI-PI is dynamic as it varies with the production characteristic of the product. It gives information about the lot number, serial number, manufacturing date, expiration date, etc.
To obtain a UDI-DI and UDI-PI code, you need to contact one of the entities the European Commission authorizes. The information that needs to be provided depends on the chosen entity.
The Basic UDI-DI and the UDI carrier
The Basic UDI-DI is an identification number specific to a group of devices with the same intended purpose, risk class, and essential design and manufacturing characteristics of a manufacturer. It is independent of the packaging or labeling of the device and does not appear on any trade item.
The Basic UDI-DI is the main access key for device-related information in the EUDAMED database and it is provided by officially designated entities selected by the European Commission. How the Basic UDI-DI is generated, the format it comes in, and the information that needs to be provided depends on the entity you choose to provide the Basic UDI-DI.
The UDI carrier is an expression of the representation of the UDI. It consists of an Automatic Identification and Data Capture (AIDC) part and a Human Readable Interpretation (HRI) part. The AIDC is a technology used to automatically capture data, including bar codes, smart cards, biometrics, and radio-frequency identification (RFID). The HRI is a legible interpretation of the data characters encoded in the UDI. Both the AIDC and HRI should contain the complete UDI information.
The UDI carrier must be placed on the label of the device and on all higher levels of packaging as well. In the case of reusable devices, it must be placed on the device itself (direct marketing). Some exceptions and more specific requirements concerning the UDI carrier can be found in Annex VI Part C Section 4 of the MDR/IVDR.
When do you have to be UDI-ready?
The UDI requirements will become mandatory once the MDR (26 May 2021) and IVDR (26 May 2022) enter into force. UDI data submission in the EUDAMED database will become mandatory on 26 November 2022, which will be 24 months after EUDAMED became fully functional. Concerning the UDI carrier, it depends on the classification of your device:
- 26 May 2021 for Class III and implantable devices
- 26 May 2023 for Class IIa and IIb and Class D devices
- 26 May 2025 for Class I and Class B and C devices
- 26 May 2027 for Class A devices
What if I’m not UDI-ready in time?
So as you can see, depending on the type of device you produce, you may not have much time left to do your UDI homework. That’s why we strongly advise you to start your UDI process in time.
In case additional support is needed, we kindly invite you to talk to our QbD experts.

with ease.
About the Author

Regulatory Affairs
Maaike specializes in Regulatory Affairs, helping life science companies navigate complex regulatory landscapes and achieve market authorization.

Navigate Regulatory Complexity with Confidence
Navigate complex regulatory landscapes with expert guidance across pharmaceuticals, medical devices, and IVD.
Read moreSubscribe to the latest updates in life science
Expert perspectives delivered to your inbox — pick your interests.
No spam, ever. Unsubscribe anytime.


