
近年来, 欧盟市场上的 医药产品 越来越多地在欧盟以外生产。虽然这一趋势在 活性成分 的生产中尤为明显 ,但在 成品药的生产中也越来越普遍 。
与此同时, 全球化 也使 药品供应链 变得 更加复杂。为确保监督,欧盟将药品进口商归类为生产商,要求他们获得生产进口授权 (MIA),并遵守《药品生产质量管理规范》(GMP) 规定。
这一框架要求实施 药品质量体系、配备足够的人员和设施、进行投诉和召回管理以及严格的供应链控制。
在本指南中,我们将探讨 GMP 附件 21的主要方面 、其对药品进口商的影响以及确保合规的最佳做法。
GMP 附件 21:概述
GMP 附件 21 规定了 MIA 持有者 从 欧盟/欧洲经济区 以外进口医药产品 的 GMP 要求。 该附件适用于
- 人用医药产品
- 兽用医药产品
- 研究用医药产品
但是,它不包括 没有欧盟/欧洲经济区上市许可但 直接再出口的产品。
GMP 附件 21中 "进口 "的定义
该附件提供了对进口的通用解释,将其定义为 从 欧盟/欧洲经济区 以外 实际引进 医药产品 。
进口是一个受监管的过程, 只有在 欧盟/欧洲经济区国家 内 进行实际进口和 清关后,质量受权人(QP)才能进行批次放行。
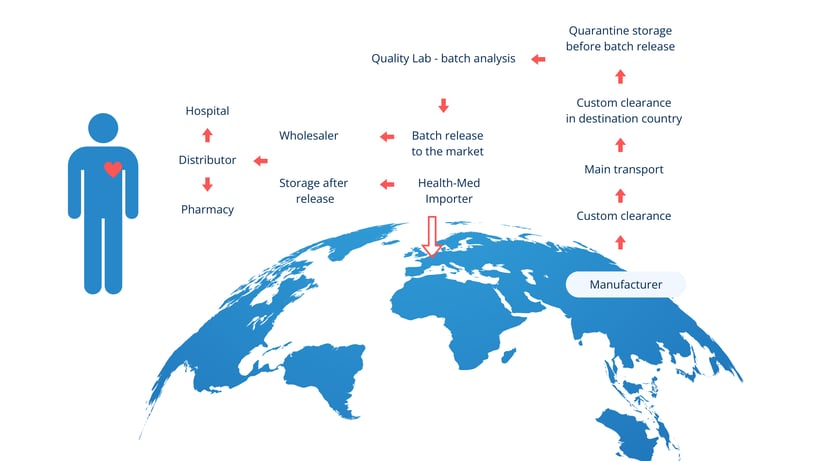
进口过程中的关键场所
GMP 附件 21 强调了涉及 医药产品进口的两个关键场所 :
- 实际进口地点 - 医药产品首次进入欧盟的地点。
- QP 认证地点 - 质量受权人 (QP) 在放行前进行批次认证的 地点 。
必须在 整个供应链的大背景下考虑这些地点 ,其中还包括
-
原料药制造商
-
第三国医药产品制造商
-
运输公司
-
进口产品的 QC 检测机构
-
营销授权持有人 (MAH)