












En el caso de los medicamentos, la información sobre el producto (PI) es un documento clave que proporciona un resumen de la información aprobada oficialmente para los profesionales sanitarios y los pacientes. La precisión y claridad del documento son cruciales, por lo que es esencial que las traducciones de la PI de los medicamentos sean precisas y adecuadas.
Aquí es donde entra en juego la Revisión Lingüística de la Agencia Europea de Medicamentos o EMA, un proceso crucial que garantiza que cada palabra de la documentación facilitada a pacientes y profesionales sanitarios de toda Europa sea precisa, coherente y cumpla los requisitos normativos.
En esta entrada del blog, ofrecemos información básica sobre la PI de los medicamentos y explicamos con más detalle el proceso y los plazos de la revisión lingüística de la EMA.
¿Qué es la información del producto (PI)?
Para los productos aprobados mediante el Procedimiento Centralizado (CP) de la UE, la Información del Producto (PI) es un documento clave que proporciona un resumen de la información aprobada oficialmente para los profesionales de la salud (HCP) y los pacientes en relación con un medicamento.
La IP comprende los siguientes documentos clave:
- El resumen de las características del producto (SmPC), que constituye la base de la información para los profesionales sanitarios sobre cómo utilizar el medicamento de forma segura y eficaz dentro de los términos de la autorización de comercialización.
- En el caso de los productos aprobados mediante el procedimiento centralizado, el anexo II contiene información sobre las condiciones o restricciones específicas de uso.
- Texto de etiquetado: se utiliza para preparar el embalaje inmediato o exterior del medicamento, como el papel de aluminio, la etiqueta del frasco o el cartón.
- El prospecto, que contiene información en consonancia con el resumen del SmPC, pero se presenta en términos sencillos adecuados para que los usuarios finales, como los pacientes o los cuidadores, puedan leerlo e interpretarlo. Se adjunta una copia impresa con el medicamento tal como se suministra para la venta.
Las versiones finales de la IP se presentan a la Agencia Europea de Medicamentos (EMA ) como un documento único que contiene todas las secciones anteriores en formato PDF, con las convenciones de formato específicas requeridas.
¿En qué consiste el proceso de revisión lingüística de la EMA?
La Revisión Lingüística de la EMA es el proceso de traducción y revisión de toda la Información del Producto (IP) nueva y actualizada requerida tras la opinión para todos los medicamentos autorizados por el Procedimiento Centralizado (PC) de la Unión Europea (UE).
El procedimiento de Revisión Lingüística de la EMA es un proceso estandarizado de 25 días que tiene lugar durante el proceso inicial de Solicitud de Autorización de Comercialización (MAA), así como tras ciertos procedimientos posteriores a la autorización que afectan a la IP, como las solicitudes de variación o la evaluación de los informes periódicos actualizados en materia de seguridad (PSUR).
Una vez emitido el dictamen del CHMP, la información del producto en inglés (EN PI) se traduce a los 25 idiomas oficiales de la UE/EEE. Estas traducciones son revisadas posteriormente por los distintos Estados miembros. Este proceso se conoce como "Revisión lingüística de la información del producto".
Las traducciones deben presentarse y revisarse según un calendario establecido que comienza inmediatamente después del Dictamen del CHMP.
- En el caso de los productos a los que se ha concedido una autorización de comercialización (MA) inicial, la fecha de la opinión del CHMP se toma como el día 210, que corresponde al final de la evaluación de la solicitud de autorización de comercialización (MAA).
- En el caso de los cambios posteriores a la aprobación, el día 0 se utiliza para definir la fecha de la opinión del CHMP.
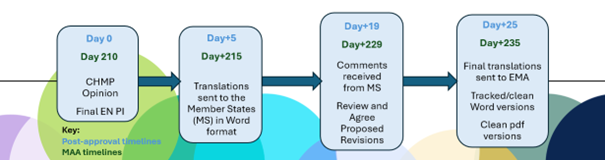
Figura 1. Plazos de la revisión lingüística y requisitos de documentación
Plazos clave del proceso de revisión lingüística de la EMA:
- Día +5 / Día +215 Presentación: Para la mayoría de los procedimientos, una vez que se adopta el dictamen del CHMP, el solicitante debe presentar las traducciones del IP solo 5 días calendario después. Dado que la reunión del CHMP concluye un jueves de cada mes, solo hay 3 días hábiles para finalizar la presentación, por lo que se requiere un enfoque pragmático para comenzar las traducciones.
- Día +25 / Día +235 Presentación: Solo hay 4 días hábiles para revisar y finalizar la presentación después de la fecha límite para los comentarios de los Estados miembros (EM) en el Día +19 / Día 229. Los comentarios de los Estados miembros pueden requerir un diálogo entre el titular de la autorización de comercialización (MAH) y el Estado miembro para llegar a un acuerdo sobre la redacción final.
Preparación, revisión y aprobación del PI
Durante la solicitud inicial de autorización de comercialización, el solicitante prepara el ES PI de acuerdo con las plantillas y los requisitos estilísticos de la Revisión de la Calidad de los Documentos (QRD).
A lo largo del proceso de evaluación, el documento se somete a múltiples rondas de revisión por parte de la agencia reguladora. En el documento se resumen todos los aspectos del expediente de CTD :
- Información sobre la seguridad y la eficacia de estudios clínicos y no clínicos, como la indicación, la posología, los efectos secundarios e información sobre poblaciones específicas, como la pediatría y las personas mayores.
- Detalles de los aspectos de calidad, como la concentración, la forma farmacéutica, los excipientes y el fabricante responsable de la liberación del lote.
- Información administrativa, como el nombre del producto, los detalles de MAH, los números MA y los tamaños de los paquetes.
En el caso de un producto centralizado, el Comité de Medicamentos de Uso Humano (CHMP) de la EMA, tras la evaluación del MAA, recomendará si la solicitud se considera aprobable.
- La opinión positiva (o negativa) se envía a la Comisión Europea (CE), que finalmente concede la autorización de comercialización de la UE, normalmente unos 67 días después de la recomendación del CHMP.
- Los mismos principios se aplican a los productos que ya tienen una autorización de comercialización: la EMA revisa los cambios posteriores a la autorización, y el CHMP adopta una recomendación sobre si el cambio es aprobable.
- Una vez más, la opinión positiva (o negativa) se envía a la CE, que actualiza los términos de la autorización de comercialización de la UE, normalmente unos 67 días después de la recomendación del CHMP o anualmente, dependiendo del tipo de cambio aplicado.
La experiencia de QbD para agilizar los procesos de revisión lingüística
A menudo, los clientes se centran, con razón, en lograr el objetivo principal de la aprobación de la solicitud de autorización de comercialización o variación, y no prestan suficiente atención al proceso de revisión lingüística de la información del producto posterior a la aprobación, que requiere mucho tiempo y recursos.
Es importante tener en cuenta que la PI será el texto utilizado para preparar el material gráfico y el etiquetado para el suministro comercial; por lo tanto, es imperativo garantizar un alto nivel de precisión y cumplimiento en todos los idiomas.
La flexible oferta de servicios integrales de QbD puede proporcionar apoyo en la preparación, revisión y presentación de las traducciones de PI, gestionando los comentarios de revisión de los EM hasta que se presenten las traducciones finales acordadas al final del proceso.
El Grupo QbD cuenta con una amplia experiencia en este ámbito. En la última década, hemos gestionado más de 250 revisiones lingüísticas. Nuestros clientes confían en nosotros para gestionar todo el proceso de revisión lingüística, y destacamos en las siguientes actividades:
- Revisar la EN PI para asegurarse de que el contenido cumpla con las pautas de estilo y formato de QRD antes de la presentación inicial de MAA o posterior a la aprobación.
- Gestionar los aspectos del proyecto, incluida la determinación de cuándo comenzar a trabajar en las traducciones iniciales para cumplir con la fecha límite de presentación. Como se ha señalado anteriormente, en el momento del dictamen del CHMP, solo hay tres días hábiles antes de la presentación para la revisión de los Estados miembros.
- Coordinar las traducciones iniciales, implementar pasos de control de calidad efectivos, entregar los archivos a los clientes para su revisión local y enviarlos para su revisión por MS de acuerdo con estrictos plazos de procedimiento.
- Utilizar procesos estándar y aplicaciones de software bien definidos para garantizar un contenido y un formato precisos, todo lo cual garantiza que cumplamos con los plazos de presentación.
- Basándonos en nuestro amplio conocimiento y experiencia con los requisitos de varios procedimientos de EMA, incluida la gestión de actualizaciones en todas las familias de productos, la distribución del trabajo y la implementación de cambios para los procedimientos autorizados centralmente y productos autorizados a nivel nacional siguiendo los procedimientos de PSUSA o de referencia.
Resumen y consideraciones futuras
La preparación de una Información del Producto (PI) en inglés de alta calidad es un punto de partida crucial para desarrollar traducciones como parte del proceso de Revisión Lingüística, que requiere mucho tiempo y recursos. Es importante señalar que la IP se utiliza posteriormente para preparar el material gráfico y el etiquetado para la producción comercial.
De cara al futuro, es probable que el desarrollo de la información electrónica sobre el producto (ePI ) afecte al proceso actual descrito en este artículo. En un próximo post, exploraremos el impacto potencial de la ePI en las agencias reguladoras, los titulares de autorizaciones de comercialización (MAH), los profesionales sanitarios (HCP) y los pacientes.








.jpg)




.jpg)

.jpg)


.jpg)
.png)