

La adopción de la inteligencia artificial (IA) en la industria farmacéutica está acelerándose, aunque a un ritmo mucho más lento que en otros sectores. ¿Por qué? En parte, porque las propiedades inherentes de los modelos de IA, y especialmente de los modelos de aprendizaje automático (machine learning), no encajaban fácilmente en el marco regulatorio existente.
El nuevo Anexo 22 tiene como objetivo cerrar esta brecha ampliando el Anexo 11 y definiendo claramente las expectativas regulatorias para la integración segura y conforme de soluciones de IA y ML en entornos bajo Buenas Prácticas de Fabricación (GMP). Este nuevo anexo detalla la posición de las autoridades en aspectos como la validación de modelos, seguimiento del rendimiento, calidad de los datos y supervisión humana.
La introducción del Anexo 22 viene acompañada de actualizaciones en:
- Capítulo 4: refuerza los controles sobre la documentación digital y la trazabilidad, y
- Anexo 11: revisa los estándares para sistemas informatizados, reflejando la integración moderna de la IA y alineándolos con el Anexo 22.
En conjunto, estas revisiones crean un modelo regulatorio armonizado y basado en el riesgo, diseñado para salvaguardar la seguridad del paciente, la calidad del producto y la integridad de los datos en una era farmacéutica impulsada por la IA.
En las siguientes secciones, analizaremos más de cerca el Anexo 22, desglosaremos sus principales disposiciones y exploraremos qué implican estas actualizaciones para las operaciones diarias de la industria.
Anexo 22: la primera guía GMP centrada en la IA
Con la introducción del Anexo 22, la Comisión Europea establece el primer marco GMP específico para inteligencia artificial en la UE, respondiendo directamente a la creciente presencia del aprendizaje automático y la toma de decisiones algorítmica en la fabricación farmacéutica.
Alcance
El anexo 22 define un alcance limitado para la aplicación de modelos de aprendizaje automático estáticos y deterministas en la industria farmacéutica, específicamente para aplicaciones críticas con un impacto directo en la calidad del producto, la integridad de los datos o la seguridad del paciente.
Desgranemos un poco más este ámbito:
- En primer lugar, esto significa que los modelos de IA para aplicaciones no críticas no necesitan cumplir los requisitos de este anexo, ni tampoco los modelos de IA que se programan explícitamente, ya que estos no se consideran aprendizaje automático, es decir, no se entrenan con datos.
- En segundo lugar, el ámbito de aplicación se limita únicamente a los modelos estáticos deterministas, lo que significa que los modelos dinámicos y/o no deterministas no están cubiertos por las normas del anexo y deben excluirse de las aplicaciones críticas de GMP.
Para aclararlo:
- Los modelos estáticos son modelos que " no adaptan su rendimiento durante el uso mediante la incorporación de nuevos datos", por lo tanto, esto cierra la puerta a cualquier sistema de aprendizaje continuo.
- Los modelos deterministas son los que "proporcionan resultados idénticos cuando se les dan entradas idénticas", lo que cierra la puerta a cualquier modelo probabilístico cuyo resultado tienda a ser impredecible.
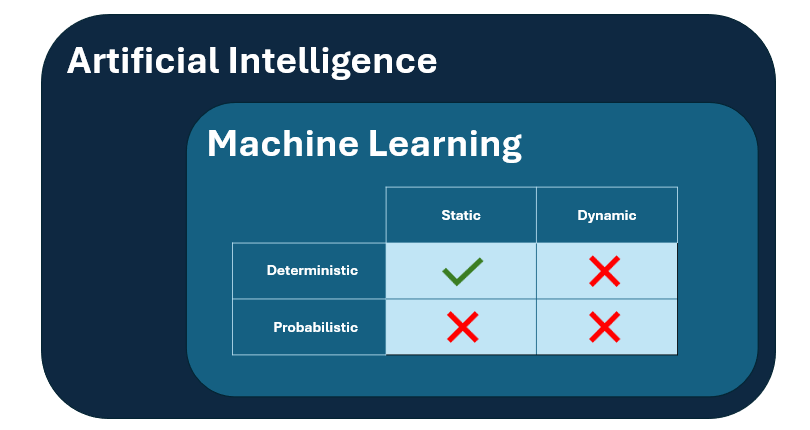
Figura 1: representación visual del subgrupo de modelos de IA que se consideran incluidos en el ámbito de aplicación del anexo 22.
Como efecto de este alcance, los modelos de lenguaje de gran tamaño, como ChatGPT y similares, no se consideran adecuados para su uso en aplicaciones GMP críticas.
Requisitos básicos
Requisitos previos al desarrollo
El anexo 22 asigna la responsabilidad de toda la documentación al usuario regulado y hace hincapié en la participación de personal cualificado durante todo el ciclo de vida del modelo.
Inicialmente, este conocimiento del proceso por parte de las PYME debe destilarse en una descripción detallada del uso previsto que debe incluir una caracterización exhaustiva del espacio muestral de los datos de entrada del modelo y una evaluación de las limitaciones y posibles sesgos de los datos de entrada.
Debe llevarse a cabo un análisis exhaustivo de este espacio muestral con una división en subgrupos si procede, ya que es importante que éstos estén suficientemente representados en los datos de entrenamiento y prueba para garantizar una buena generalización del modelo y evitar un rendimiento inferior para determinadas partes del espacio muestral de entrada.
Además, debe definirse claramente la participación de un usuario humano en el proceso (human-in-the-loop) y su responsabilidad al interactuar con un modelo de IA.
Antes de realizar cualquier prueba de aceptación, deben definirse y aprobarse métricas de prueba y criterios de aceptación adecuados para el rendimiento del modelo, teniendo en cuenta que el modelo que se va a implantar debe tener, como mínimo, el mismo rendimiento que el proceso actual al que sustituye.
A partir de ahí, el anexo no habla del proceso de selección y formación del modelo y se centra en los datos de prueba y los requisitos de las pruebas.
Requisitos de las pruebas de aceptación
Preparación de pruebas
Las pruebas deben planificarse y documentarse en un plan de pruebas aprobado que incluya:
- Resumen del uso previsto
- Métricas predefinidas y criterios de aceptación
- Referencias a datos de prueba y scripts
- Descripciones paso a paso de las pruebas y del cálculo de métricas
- Cualquier desviación debe documentarse, investigarse y justificarse
Calidad de los datos de prueba
El anexo hace un énfasis notable en la importancia de la “calidad” de los datos de prueba, y con razón. En resumen, un conjunto de datos de prueba puede considerarse de alta calidad si cumple con todos los puntos de la siguiente lista:
- Cubre todo el espacio muestral definido en la descripción de uso previsto y es representativo de los datos de entrada del entorno real de uso, incluyendo cualquier preprocesamiento de datos.
- Contiene datos suficientes para todos los subgrupos del espacio muestral, de manera que se puedan estimar con confianza los indicadores de prueba predefinidos.
- Está correctamente etiquetado, lo cual debe verificarse de forma rigurosa.
- Es completamente independiente, lo que significa que los datos utilizados para evaluar el desempeño del modelo no influyen de ninguna manera en su desarrollo, selección, entrenamiento o validación.
- Cualquier limpieza, exclusión o generación artificial de datos de prueba se realiza únicamente cuando es estrictamente necesario y queda debidamente documentada y justificada.
Independencia de los datos de prueba
El acceso a los datos de prueba debe ser controlado y trazable, con registros que indiquen quién accedió, cuándo y con qué frecuencia.
El personal involucrado en el entrenamiento del modelo no debería tener acceso a estos datos; si fuera inevitable, debe implementarse un sistema de doble revisión, donde una persona sin acceso previo acompañe a quienes sí han visto los datos.
Confianza y explicabilidad
La aceptación de un modelo debe considerar tanto la explicabilidad como la confianza en sus resultados. El anexo espera que el sistema sea capaz de identificar las características principales de los datos de prueba que influyen en las decisiones del modelo, revisando su relevancia.
Además, los modelos de predicción o clasificación deben registrar las puntuaciones de confianza, que reflejan la certeza y fiabilidad de los resultados. Debe establecerse un umbral apropiado de confianza para marcar como “indecisos” aquellos resultados que no lo superen.
Requisitos operativos
El anexo establece requisitos específicos para el uso operativo de los modelos de Machine Learning. Por ejemplo, el modelo debe estar bajo control de configuración, evitando y detectando cualquier cambio no autorizado.
Todo el sistema y proceso en el que se implementa el modelo debe estar bajo control de cambios. Cualquier modificación en el modelo, sistema, proceso, datos de entrada o entorno debe documentarse y evaluarse por su impacto en el desempeño del modelo. En estos casos, la revalidación debería ser la acción predeterminada, y cualquier decisión de omitirla debe justificarse adecuadamente.
El rendimiento de un modelo también puede degradarse sin cambios deliberados, debido a variaciones ambientales o cambios no planificados en el espacio muestral; por ello, el desempeño debe monitorearse continuamente con respecto a los indicadores predefinidos.
Si el modelo se utiliza como input en decisiones humanas (“human-in-the-loop”), y esto se usa para limitar las pruebas del modelo, todo el proceso debe registrarse y supervisarse minuciosamente.
Integración armonizada: Un marco unificado
La verdadera fortaleza de las revisiones 2025 de Eudralex no reside solo en el contenido del Anexo 22, sino en cómo el Anexo 22, el Capítulo 4 y el Anexo 11 se interconectan para formar un marco regulatorio coherente y preparado para el futuro.
Capítulo 4 – Documentación
El Capítulo 4 se ha actualizado para reflejar la realidad digital de las operaciones GMP:
- Nuevas disposiciones para la captura automática de datos y generación de registros, especialmente mediante herramientas habilitadas con IA.
- Mayor énfasis en trazabilidad y auditabilidad, garantizando que los registros digitales o influenciados por IA cumplan plenamente con los principios ALCOA+.
- Integración de registros electrónicos dentro del sistema documental, incluyendo control de versiones, seguimiento de metadatos y acceso seguro.
Anexo 11 – Sistemas informatizados
El Anexo 11 regula los sistemas informatizados en entornos GMP y se ha actualizado para reflejar la sofisticación de herramientas modernas basadas en IA y en la nube:
- Alineación clara con el Anexo 22, considerando la IA como un subconjunto de sistemas informatizados, pero con requerimientos de control y validación específicos.
- Refuerzo de la integridad de los datos, especialmente en sistemas híbridos donde la entrada humana y de IA se combinan.
- Actualización de los requisitos sobre firmas electrónicas, seguridad de sistemas, copias de seguridad y registros de auditoría.
Estos cambios posicionan al Anexo 11 como la columna tecnológica que soporta las directrices específicas del Anexo 22, creando un marco de cumplimiento más integral y moderno.
Juntos, crean un marco normativo coherente y basado en el riesgo que regula activamente el uso de la IA en entornos de PCF en lugar de limitarse a tolerarlo.
Esta integración adapta la carga reglamentaria al impacto del sistema de IA en la calidad de los productos, la integridad de los datos y la seguridad de los pacientes: un enfoque moderno y flexible adaptado a las tecnologías digitales emergentes.
Impacto práctico para las partes interesadas del sector farmacéutico
Con estos cambios ya formalizados, las diferentes partes interesadas del ecosistema farmacéutico tendrán que adaptarse no solo a las nuevas normas, sino también a una nueva mentalidad en torno a la IA y los sistemas digitales en entornos GMP.
Para fabricantes y equipos de calidad
- Establecer marcos de gobernanza de IA que incluyan selección, validación y monitoreo de modelos.
- Adaptar procesos de validación a sistemas de IA iterativos o no determinísticos.
- Fortalecer evaluaciones de riesgo, control de cambios y supervisión humana.
Para profesionales de Reguatory Affairs y Compliance
- Prepararse para mayores expectativas sobre integridad y trazabilidad documental.
- Asegurar que los registros relacionados con IA cumplan estándares completos de auditoría.
- Mejorar la colaboración transversal entre QA, IT y equipos de data science.
Para proveedores tecnológicos y desarrolladores de IA
- Diseñar sistemas alineados con validación GMP y control del ciclo de vida.
- Proveer herramientas de transparencia y explicabilidad.
- Demostrar conocimiento de los requerimientos del Anexo 22 como ventaja competitiva en selección de proveedores.
En resumen, la conformidad ya no es sólo cuestión de documentación, sino de diseño. Los sistemas de IA deben diseñarse desde cero para garantizar la trazabilidad, la rendición de cuentas y la auditabilidad.
Conclusiones
La introducción del Anexo 22 supone algo más que una actualización normativa; señala un cambio cultural en la forma en que la industria farmacéutica aborda el aprendizaje automático en procesos críticos.
Atrás quedaron los días de tratar la IA como una "caja negra". La Comisión Europea lo ha dejado claro:
Si la IA influye en la seguridad del paciente, la calidad del producto o la integridad de los datos, debe ser transparente, controlada por sesgos, validada, trazable y supervisada continuamente.
Estas directrices no reprimen la innovación: la canalizan de forma responsable, estableciendo expectativas claras y basadas en el riesgo que permiten a las empresas innovar con confianza, manteniendo al mismo tiempo los más altos estándares de calidad.
A medida que otros organismos reguladores de todo el mundo miran a la UE en busca de liderazgo, el anexo 22 podría convertirse en un punto de referencia mundial para la gobernanza de la IA en entornos de buenas prácticas de fabricación. Las empresas que se preparen con antelación, alineando sistemas, documentación y prácticas de calidad, estarán mejor posicionadas para el cumplimiento y el liderazgo en la próxima era de la fabricación farmacéutica.
¿Preparado para el Anexo 22: 2026? Preparemos su estrategia de conformidad para el futuro.
No esperes a la publicación final; empieza a prepararte hoy mismo. Colabora con QbD Group para evaluar tus sistemas, validar los modelos de IA y crear una estrategia de cumplimiento a prueba de futuro que mantenga a tu organización preparada para las inspecciones.
¿Necesitas ayuda para navegar por la revisión del Anexo 22?
Ponte en contacto con nuestros expertos en conformidad y validación.

¿Buscas garantizar la conformidad y fiabilidad de tu software?
Desde la gestión de datos hasta la validación del software, nuestras soluciones garantizan el cumplimiento normativo y la eficiencia operativa.
Sobre el autor

Life Science Consultant at QbD Group
Ward is a Life Science Consultant at QbD Group, providing expert guidance on quality, regulatory, and compliance challenges in the pharmaceutical industry.
QbD Group
¿Listo para acelerar tu proyecto en Life Sciences? Habla con nuestros expertos.
Contacta con un experto →

