
Requisitos de conformidad para los importadores
En el caso de los medicamentos importados de países sin Acuerdo de Reconocimiento Mutuo (MRA), la legislación de la UE exige la realización de pruebas por lotes en el momento de la importación, antes de la certificación de la garantía de calidad y de la liberación, de conformidad con el anexo 16 sobre prácticas correctas de fabricación.
El lugar de las pruebas y los requisitos mínimos de las mismas se describen en:
- la letra b) del apartado 1 del artículo 51 de la Directiva 2001/83/CE (para los medicamentos de uso humano)
- Artículo 55(1)(b) de la Directiva 2001/82/CE (para medicamentos veterinarios).
Referencia a las orientacionesde Eudralex
El anexo 21 de BPF hace referencia a otros capítulos y anexos de Eudralex, en particular al anexo 16, que ya define las responsabilidades de una persona cualificada (PC) para la certificación y liberación de lotes.
Sin embargo, el Anexo 21 enfatiza aún más el papel del Titular de la Autorización de Comercialización (TAC) para garantizar el cumplimiento de las BPF durante todo el proceso de importación y fabricación.
Por ejemplo:
-
El MAH debe asegurarse de que los QP tengan pleno acceso a la documentación del lote en todo momento.
-
El titular de la autorización tiene la responsabilidad última de la comercialización de los medicamentos y debe tener acuerdos por escrito con todos los centros pertinentes.
Revisión de la calidad del producto (PQR) en la importación
El Anexo 21 también aclara los requisitos de la Revisión de la Calidad del Producto (PQR) para los productos importados. La QP certificadora debe:
-
Comparar los resultados analíticos del centro de pruebas de importación con el certificado de análisis del fabricante del tercer país.
-
Garantizar la existencia de un programa de estabilidad permanente, incluso si las pruebas se realizan en un tercer país, siempre que la QP tenga acceso a los datos de estabilidad.
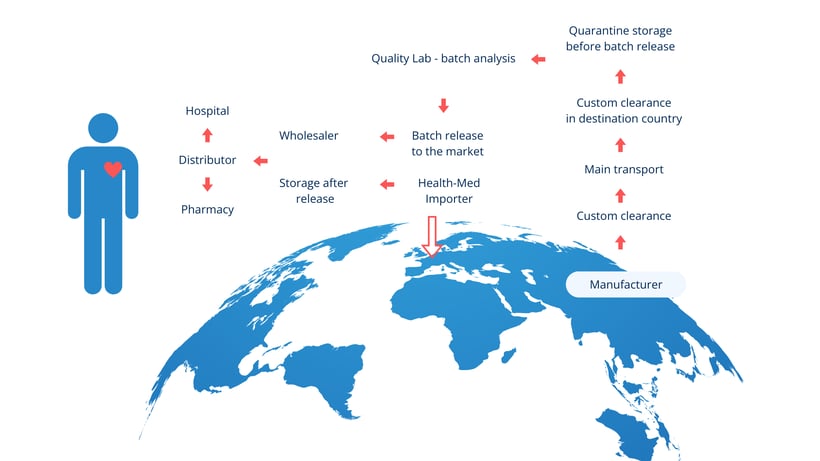
Conclusión
A medida que la fabricación de productos farmacéuticos y las cadenas de suministro siguen globalizándose, el GMP Anexo 21 desempeña un papel crucial a la hora de garantizar la supervisión regulatoria de los medicamentos importados.
El anexo refuerza el marco para los titulares de MIA, haciendo hincapié en las responsabilidades de las QP, la integridad de la cadena de suministro y un riguroso control de calidad.
Las empresas que importan medicamentos a la UE deben anticiparse a los requisitos de cumplimiento, garantizar una documentación sólida y establecer colaboraciones sólidas en toda la cadena de suministro.
¿Necesita ayuda para cumplir las GMP en la importación de medicamentos?
En QbD Group ofrecemos servicios integrales a las empresas que importan medicamentos a la UE. Nuestro laboratorio con certificación GMP y nuestros servicios de persona cualificada (QP) garantizan la liberación de lotes sin problemas , el cumplimiento de la normativa y unos procesos de importación eficientes.
Simplificamos tu acceso al mercado de la UE. Ponte en contacto con nuestros expertos hoy mismo.

¿Necesitas servicios de servicios de laboratorio y de validación en los que puedas confiar?
Sobre el autor

MP, QP · Division Head Lab Services
Yves leads the Lab Services division at QbD Group, bringing deep expertise in GMP laboratory operations, analytical method validation, and quality control for pharmaceutical manufacturing.
QbD Group
¿Necesita ayuda con los procesos regulatorios? Nuestros expertos pueden guiarle.
Solicitar asesoría experta →Suscríbete a las últimas novedades en Life Sciences
Perspectivas de expertos en tu bandeja de entrada — elige tus intereses.
Sin spam, nunca. Cancela cuando quieras.


