

En la industria farmacéutica, un Sistema de Gestión de la Calidad (QMS) es la columna vertebral de todas tus actividades de cumplimiento y aseguramiento de la calidad. Ya sea en la gestión de desviaciones, CAPAs, auditorías, formación o documentación, tu QMS soporta directamente procesos clave que impactan en la calidad del producto y la seguridad del paciente.
Si tu QMS está basado en software, debe estar validado. Es decir, debes demostrar, con evidencia documentada, que el sistema funciona según lo previsto y lo hace de forma consistente en el tiempo.
Este no es solo un buen ejercicio de control interno, es un requerimiento regulatorio: las autoridades como la FDA y la EMA exigen que cualquier sistema informatizado empleado en procesos GxP (Good Manufacturing Practice, Good Clinical Practice, etc.) esté correctamente validado.
En este artículo profundizaremos en qué significa validar un QMS, por qué es crítico y cómo hacerlo de forma eficaz basándonos en las guías clave: Anexo 11 de EU GMP, 21 CFR Parte 11 y GAMP 5.

¿Buscas garantizar la conformidad y fiabilidad de tu software?
Desde la gestión de datos hasta la validación del software, nuestras soluciones garantizan el cumplimiento normativo y la eficiencia operativa.
Por qué es importante la validación
La garantía de software (Computer Software Assurance, CSA) asegura que tu sistema ofrece un rendimiento fiable, produce datos íntegros y soporta los procesos críticos de calidad y compliance. Si tu QMS controla desviaciones, SOPs o procesos CAPA, un fallo en el sistema puede afectar directamente el cumplimiento normativo o la calidad del producto.
Sin validación:
- Los flujos de trabajo críticos podrían fallar
- Las desviaciones o CAPAs podrían gestionarse incorrectamente
- Los registros documentales o de formación podrían quedar incompletos
- El cumplimiento normativo y la calidad se verían comprometidos
La validación de tu QMS garantiza:
- Comportamiento esperado del sistema
- Registros electrónicos seguros y trazables
- Auditorías e integridad de firmas electrónicas conforme a normativa
- Datos fiables para la toma de decisiones
No es un mero trámite regulatorio. Es un pilar clave de la gestión de riesgo, la integridad de datos y la protección de la operación y el paciente.
Entendiendo el entorno regulatorio
Existen varias guías clave que definen qué se espera en la validación de sistemas informatizados, y aplican de manera directa a la validación de QMS:
- EU GMP Anexo 11: Sistemas informatizados
- 21 CFR Parte 11 (FDA): Registros y firmas electrónicas
- GAMP 5: Guía práctica para validación basada en riesgo
- ICH E6(R3): Buenas prácticas clínicas para sistemas en ensayos clínicos
- ISO 13485: Requisitos del QMS para dispositivos médicos
- EU MDR / IVDR: Requisitos de software en dispositivos de diagnóstico
💡 Consejo : No todos los sistemas están sujetos a las mismas regulaciones. Algunos procesos pueden requerir solo la norma ISO 13485, mientras que otros que apoyan las GMP o las actividades clínicas también deben cumplir con el Anexo 11 y 21 CFR Parte 11. Céntrate en el alcance y el uso previsto de tu QMS, y aplica los marcos pertinentes a los procesos de GxP que tu QMS apoya.
¿¿Deben validarse todos los QMS?
Sí. Si el QMS se utiliza para procesos de GxP, debe validarse.
Muchos proveedores ya realizan pruebas exhaustivas y ofrecen documentación de soporte (lo que se conoce como validación de proveedor), que incluye:
- Especificaciones funcionales
- Registros de pruebas de instalación
- Informes de validación del sistema
A menudo se denomina validación del proveedor. Aunque no sustituye tu responsabilidad, puede reducir considerablemente el esfuerzo. Puedes reutilizar o hacer referencia a la documentación del proveedor para apoyar tu propio proceso de validación, especialmente con un enfoque basado en el riesgo.
Recuerda : la validación consiste en garantizar que el sistema funciona para el uso previsto, en su entorno. Aunque el proveedor valide tu plataforma, tú eres responsable de asegurarte de que tu configuración específica es adecuada para el uso previsto.
¿Qué hace diferente la validación de un QMS?
A diferencia de otros sistemas como un ERP o un MES, un QMS no gestiona transacciones o fabricación, sino procesos de calidad.
Una validación adecuada debe confirmar que el sistema garantiza de forma consistente:
- Control documental (versionado, obsolescencia, aprobación)
- Firmas electrónicas seguras, trazables y conformes
- Flujos de trabajo CAPA, desviaciones o cambios con rutas y aprobaciones correctamente definidas
- Gestión de datos maestros (categorías de documentos, tipos de eventos, plantillas)
- Trazabilidad (ej. vinculación desviaciones-CAPA, SOPs-formación)
- Control de accesos basado en roles
- Auditorías automáticas, íntegras y a prueba de manipulaciones
Estas funciones son fundamentales para un QMS, y un fallo en cualquiera de ellas puede comprometer el cumplimiento o la calidad del producto.
Cómo validar tu QMS: un enfoque práctico
Analicemos un enfoque simplificado, aunque conforme con la normativa, para validar un QMS. En lugar de una lista de comprobación de validación completa, destacaremos las áreas críticas y los pasos prácticos para garantizar que tu QMS es compatible con los procesos de calidad del mundo real.
Dependiendo de la categoría GAMP del sistema, pueden ser necesarias diferentes actividades de prueba, pero aquí nos centraremos en las áreas funcionales críticas específicas de un QMS.
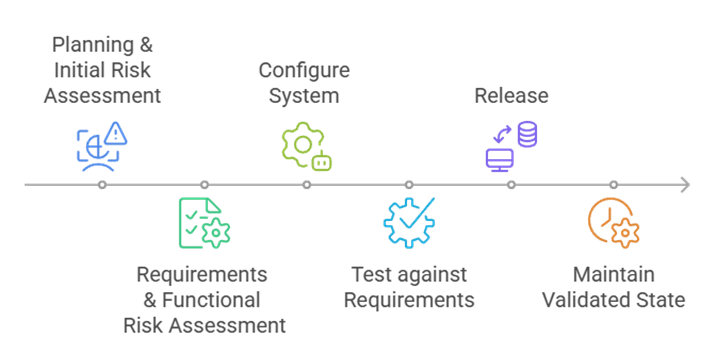
1. Planificación y análisis de riesgos
Comienza con un Plan de Validación que incluya:
- Definición del alcance, objetivos, roles y responsabilidades.
- Enfoque basado en riesgos, conforme a GAMP 5.
- Identificación de procesos críticos como desviaciones, CAPAs, formación y gestión de cambios.
- Categorización del sistema (la mayoría de los QMS son categoría 4 – configurables).
Tip: El esfuerzo de validación debe ser proporcional a la complejidad. Plataformas altamente personalizadas requieren más testing y documentación; sistemas ampliamente configurables necesitarán menos.
2. Definición de requisitos y priorización de riesgos
Elabora un URS (User Requirements Specification) centrado en tu flujo de trabajo, no en funcionalidades estándar o genéricas.
Ejemplos:
- "Debe garantizar control de versiones y prevenir el uso de SOPs obsoletos."
- "Una CAPA no puede cerrarse sin análisis de causa raíz y verificación de efectividad."
- "La revisión de un SOP debe asignar automáticamente formación a todo personal afectado."
Luego, realiza una evaluación funcional de riesgos considerando:
- Relevancia GxP: Procesos como CAPA, change control o training pueden ser de alto riesgo.
- Severidad y probabilidad de fallo.
- Funcionalidad configurable vs. personalizada: Integraciones o reportes complejos requieren mayor profundidad en el testing.
Documenta la configuración en un Configuration Specification (CS) con detalle funcional sobre:
- Workflows, roles y permisos.
- Datos maestros: tipos de documentos, eventos, plantillas.
- Lógicas de vinculación (p. ej., desviación → CAPA).
3. Configuring Your System
Con los requisitos definidos, el siguiente paso es configurar el QMS en base a ellos. La mayoría de los sistemas modernos permiten alta configurabilidad sin desarrollo a medida.
Posibles configuraciones:
- Flujos de CAPA, desviaciones o change control.
- Campos obligatorios para datos críticos.
- Roles, permisos y notificaciones.
- Vínculos entre registros relacionados.
Tip: Guarda evidencia de la configuración (screenshots, exportaciones, logs) para soporte de trazabilidad y auditorías.
4. Testing del sistema – Enfoque en áreas funcionales críticas
La validación debe demostrar que el QMS soporta tus procesos de calidad de forma consistente. El esfuerzo de testing (unit, functional, UAT) dependerá de la categoría GAMP del sistema.
Áreas de testing clave en QMS:
- Control de versiones de documentos y gestión de obsoletos.
- Firmas electrónicas seguras (21 CFR Part 11 / Annex 11).
- Rutas y aprobaciones en workflows (CAPAs, cambios, eventos).
- Relación automática entre objetos (desviación ↔ CAPA).
- Notificaciones y alertas en los puntos adecuados.
- Campos obligatorios que no deben omitirse.
- Creación rápida de registros con sellos de tiempo.
- Asignación automática de formación según revisión de SOPs.
- Métricas fiables de calidad (CAPA aging, desviación trends).
- Informes para auditorías y revisiones de gestión.
- Integraciones con otros sistemas (ERP, DMS, ALM).
Tip: Cada test debe trazarse al URS y al CS, demostrando cobertura funcional y cumplimiento.
5. Liberación del sistema
Una vez completado el testing:
- Documenta resultados en un Validation Summary Report.
- Forma a los usuarios en workflows validados.
- Implementa control de cambios y revisiones periódicas.
- Actualiza SOPs donde sea necesario.
6. Mantenimiento del estado validado
La validación es un proceso continuo. Implementa medidas para asegurar que el sistema se mantiene en estado controlado:
- Change control: Evaluación de impacto de cambios y revalidación, cuando aplique.
- Periodic review: Confirma que el sistema sigue funcionando como se espera y responde a las necesidades actuales.
- Incident management: Registro y tratamiento de incidencias o desviaciones del sistema.
Controles críticos de conformidad
Como parte de tu validación, asegúratede que el QMS incluye las siguientes características para cumplir las expectativas normativas:
- Audit trail: Registro automático, completo y seguro de cambios.
- Firma electrónica: Atribuible, segura y conforme a CFR/Annex 11.
- Acceso basado en roles: Gestión clara de permisos y usuarios.
- Data integrity ALCOA+: Atributable, legible, contemporáneo, original, preciso, completo, consistente, perdurable y accesible.
Conclusión
La validación de un QMS puede parecer un ejercicio técnico, muy orientado a IT, pero constituye un pilar crítico dentro del sistema de calidad farmacéutico. Incluso si no eres especialista en validación, comprender los fundamentos te permite mitigar riesgos, evitar hallazgos en auditorías y garantizar una toma de decisiones fiable.
Puntos clave a tener en cuenta:
- Si tu QMS da soporte a actividades GxP, eres responsable de validarlo.
- Aprovecha la documentación del proveedor siempre que sea posible, pero adapta la validación a tu uso previsto.
- Aplica un enfoque claro y basado en riesgo, alineado con Annex 11, 21 CFR Part 11 y GAMP 5.
- Prioriza tus propios flujos de trabajo; la validación no es un ejercicio documental, sino la garantía de que el sistema soporta de forma eficaz y fiable tus procesos reales.
Con la estrategia adecuada, la validación deja de ser una carga para convertirse en una inversión en confianza, control y compliance.
Si necesitas orientación o soporte operativo, en QbD podemos ayudarte. Desde la planificación y la documentación, hasta el testeo y el mantenimiento del estado validado, aportamos la experiencia necesaria para que tú puedas centrarte en entregar productos seguros y de alta calidad.
¿Necesitas apoyo para validar tu QMS? Contacta con nosotros y conversemos sobre cómo podemos acompañarte.
Sobre el autor
QbD Group
¿Listo para acelerar tu proyecto en Life Sciences? Habla con nuestros expertos.
Contacta con un experto →

